It is evident to me that images can carry and convey truth – small and capital T – and that most people aren’t actually talking about truth when they talk about objectivity and subjectivity. Strong statement, I know, but I think as usual, the people using the Internets simply bring their own ideas and not really paying attention to how writers define their terms.
Before we begin, I just want to say that nothing I speak of here is new. What I aim to provide is a new context that might help reach a new point of view. If what I say is earth-shattering to you, fabulous! If I simply reminded you of what you know, great! If you are bored by what I’m saying because it is all self-evident to you, then just simply count me as a fellow traveler.
Photographers interested in creating art are too busy creating art to let us know what they think. I absolutely agree with that; and those photographers who are interested in propagating what they have learned are working deeply, via books. One recommendation is On Being a Photographer, by Magnum photographer David Hurn and Bill Jay. The book is a dialogue between the two, and the first chapter already dives deep into concepts of art, expression, truth, and depiction via photography. I highly recommend this book; sure, my thoughts and theirs on photography align, but the book is clear and concise. They are also humane and do not condescend to non-professionals. This is only one of many books that dive deep into photography. My point – and I admit I buried it unduly in a lot of text – is that online discourse generally isn’t geared to deep analysis: why else would TL;DR be an apologia and an excuse?
OK, back to T/truth. The last thing this world needs is to have only “experts” decide on deep matters. Truth should simply matter to us as a condition for human dignity in the context of society. I know of what I speak: I was an “expert”, working in academic science, using funding from the National Institute of Health (NIH) in the United States to perform basic science research. My mentality is actually the opposite of the kind of academic who finds it beneath contempt to even consider talking to the proles. I believe in engaging non-practitioners, whether it be with the technologies I’m building or the science I am doing. Speaking to other academics is only a part of my job and interest; as your eyeballs could attest, I would write until they dry out!
Here’s what I know about truth: at some level, it is a descriptor of things that happened. In scientific articles, we present a section devoted to methods. The idea is that anyone can use that as a plan to reproduce the same conditions that led to observations. Here’s the key: you might still differ in your interpretation of the results, but the “truth” here is the baseline observation. When you do this, that happens.
There is a reason why we have difficulty imagining photography as something other than a documentary device; in all other forms of art, humans had aimed to achieve something close to photorealism. Once we had done that, we moved on to more impressionistic and abstract approaches to art, aimed to achieve emotional connection via resonance. What do you do with photography in this context? We use the term photorealistic as a way of praising artistic skill; needless to say, photography has its own baggage; its problem is precisely because it is “photorealistic”!
As others have noted, photographers own their frame. That is, they decide what goes in there. Even in a “straight out of camera” shot, much artifice can be accomplished with lighting, make-up artists, set designers, perspective, lens choice, and so forth. I’ve seen fashion model shots that are pretty much visually perfect even before retouching with Photoshop. And even with film, there’s much that can happen when the print is made.
If we can routinely fool our eyes (and with video, we can fool our eyes and ears), what can I possibly mean by truth existing, let alone that there is such a thing as objectivity?
Well, as a scientist, I used a lot of imaging techniques. I had wanted to include a lot more, but decided to cut down and present a familiar looking technology before diving off the deep end in a subsequent post.
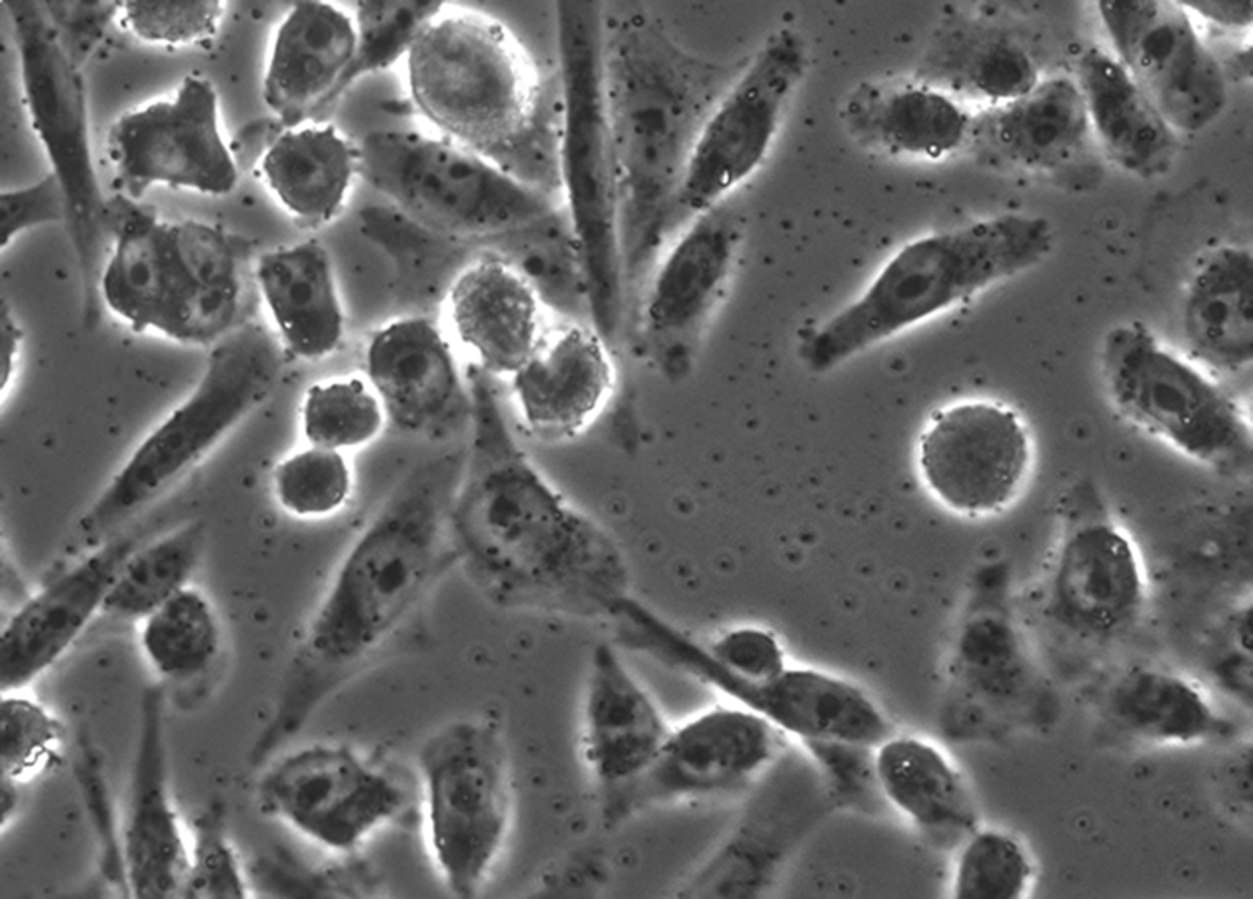
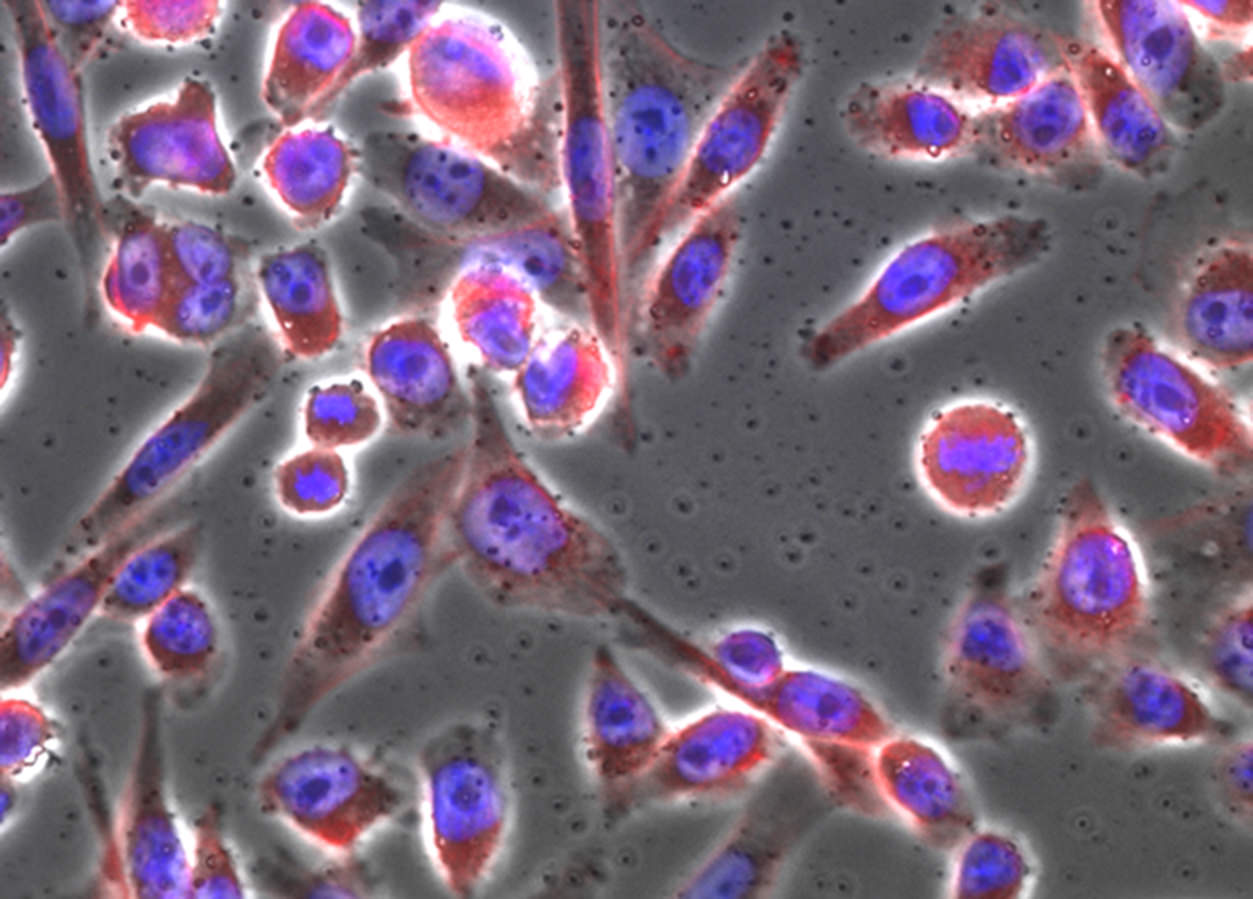
The above image is something that anyone of us may have seen: it’s a microscope image of cells. For the record, these are Chinese Hamster Ovary cells. These are “immortalized”, which is to say they continue to grow and divide for as long as they are maintained in a nutrient bath. The image was taken using a phase microscope. The cells were first grown on glass coverslip. Once stable, we can “kill” and “fix” the cells by adding a liquid call formaldehyde. This chemical reacts with nearly all molecules in the cells, seizing up the molecular machinery that allows cells to live. Chemical reactivity shuts down. Because formaldehyde lowers reactivity, it prevents decay and degradation to some extent. The cell is still exposed to compounds that degrade it, but because it is fixed, the cells no longer take part in any chemical reaction. Decay will take a lot more time, but in that span, scientists can do things like add coloring agents or image the cells.
The process of fixing and placing the cells under glass is called mounting; once mounted, we can simply view it under a microscope. The simplest microscopes are compound microscopes: the slide is essentially backlit, and the imaging objective (lens) is aligned precisely to the light source. The lens technology is a little bit specialized; it’s designed to emphasize differences in light paths arriving light collector (it could be the eye, or the CCD camera). The object, in this case the cell, refracts light as it passes through the cell. This essentially slows light down and leads to light arriving at the light collector at different times (the light is out of phase). Using phase differences to generate visual contrast allows for scientists to visualize the cell and its organelles. Without phase contrast, we would be hard pressed to even see the cells – as the differences in absorption and refraction would be undetectable to the eye. The cell might be a ghostly image at best.
Other ways of introducing visual contrast is to simply color the cell.
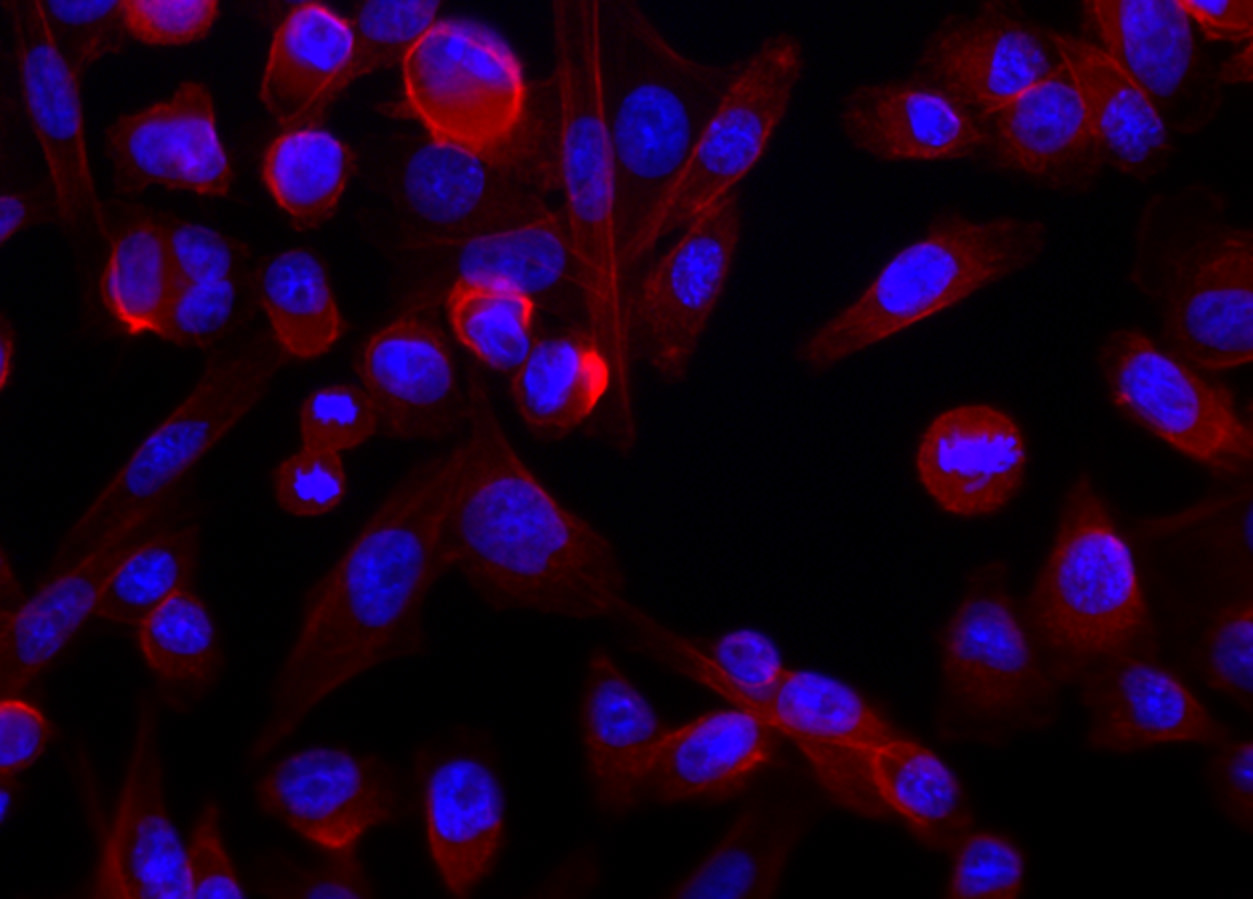
The lead image shows the same cells lit up using epifluorescence microscopy. The idea is simple: we can use coloring agents to identify structures within cells. Using fluorescence imaging lets us easily use multiple agents. And the reason for that is because dyes have excitation and emission characteristics; they are made to enhance these properties. Usually, these dyes are listed with a nominal peak excitation and emission wavelengths. The peaks, taken by themselves, would be a nominal color (e.g. 480 nm is blue; 510 nm would be green). Realistically, however, there is a range of wavelengths that can excite the dye, and each dye emits a relatively broad band of light.
What do I mean? Light can be described by wavelengths; visible light, for humans, fall within 400 to 700 nm (going from violet to blue to green to yellow to orange to red). Shorter wavelengths also correspond to more energy. This is a natural process where we can’t get more energy out than what we put in; so if we were able to excite a fluorescent dye with blue light, we’d get something with a longer wavelength than that (green, or yellow, or red). That’s actually the classic property of Green Fluorescent Protein (GFP), which has a nominal excitation peak of 480 nm and an emission peak at 514 nm.
The neat thing about dyes is that some of them are naturally selective. Rather than binding – we usually use the term, “label” – everything indiscriminately, they can target things like nucleic acides, specific proteins, fats that constitute the membranes of cells and organelles, and so forth. The blue you see is a dye called Hoechst 33342. It binds DNA preferentially. Some dyes bind all nucleic acids, but their chemistry actually allows them to exhibit different fluorescent properties depending on whether they bind RNA or DNA. Still other labels bind a protein called actin; if you attach a dye to that molecule, then you have a specific dye. In the image, it is fluorescing red.
The camera we used is a CCD camera; there is no Bayer filter in place; the camera records in 12-bit grayscale. The amount of fluorescence is actually quite low, compared to the number of excitation photons, not to mention the total output from our lamp. There is no ambient light, as it would be too bright (i.e. we kill the room lights). The cooling part is important because we reduce thermal noise. Dark noise is the native, errant filling of pixels in the absence of light. Shot noise is the variance in the performance of the pixel to a constant amount of light stimulation. Lowering these noise types will give us cleaner data. The nice thing is, because everything is static (we are talking about a tolerance of less than a 0.001 mm), it’s trivial to capture multiple images of the same scene. With multiple frames, we can average out the noise at the pixel level, resulting in clean signals, even in the absence of getting more separation between the fluorescence and background. We can do both, of course; minimize noise and expose for our signal. Hoechst 33342 happens to be a fairly strong dye. A small quantity will give a strong signal, after even a 50 ms exposure. I can tell you that most dyes do not perform like that and require 5 seconds (100 times more light, or, nearly 8 stops worth) or more.
Generally, the light is mostly broadband, and so we can cut it down using filters in front of the light source. Likewise, because the sensor itself has no color filters, we need to add a filter in front of the color, to allow only the color of light we wish to visualize.
So that’s pretty much the mechanics of the system. We see the blue in the image, which is localized to the nucleus of the cell. In the nucleus resides DNA; we need UV light (350 nm) and we read out at around 450 nm. For actin fibers, we used a red dye (excitation at 594 nm/emission at 610 nm). That protein is scaffolding that gives the cell its shape, and so it’s everywhere in the cytoplasm.
The lead image and the image with only DNA and actin stains, are simply composites that generated by the microscope’s acquisition software. These are easy enough to stack and overlay in Photoshop. The other minor point is that scientific imaging requires true pixel values. We keep images in TIFF format, with lossless compression. With the use of calibration markers, we might be able to convert the signal into actual quantities of interest. However, this is difficult, due to the reciprocity of the dye. For a host of reasons (dye access, reaction proportion, whether the cell was well fixed, etc.), dimness or brightness is not directly related to number of actual molecules that might be in the cell..
OK, so what does all this have to do with the truth? Well, I and other scientists can use images to demonstrate things. At its simplest, this type of picture is a beginner’s image. Anyone who buys these stains will want to test them out. So we use the dye, in different concentrations, on this and other cell types. In general, we follow recipes and cells are labeled and we can take pictures. I have no financial stake in the success of the dye; none of what I’ve shown you is new knowledge, but I can reproduce it all the same.
Simply demonstrating that I can stain cells and take proper pictures is only the first building block. I can then proceed to add compounds that aim to force DNA or actin into different configurations (for example if I were interested in drugs that can affect tumor cells).
What if I told you that we can capture images of cells with UV light? Not a big deal, right? What if I told you that we can process these images to make maps of nucleic acid and protein distribution? What if I told you that we can simply sum up the pixel values to give us a total mass of each class of molecules?
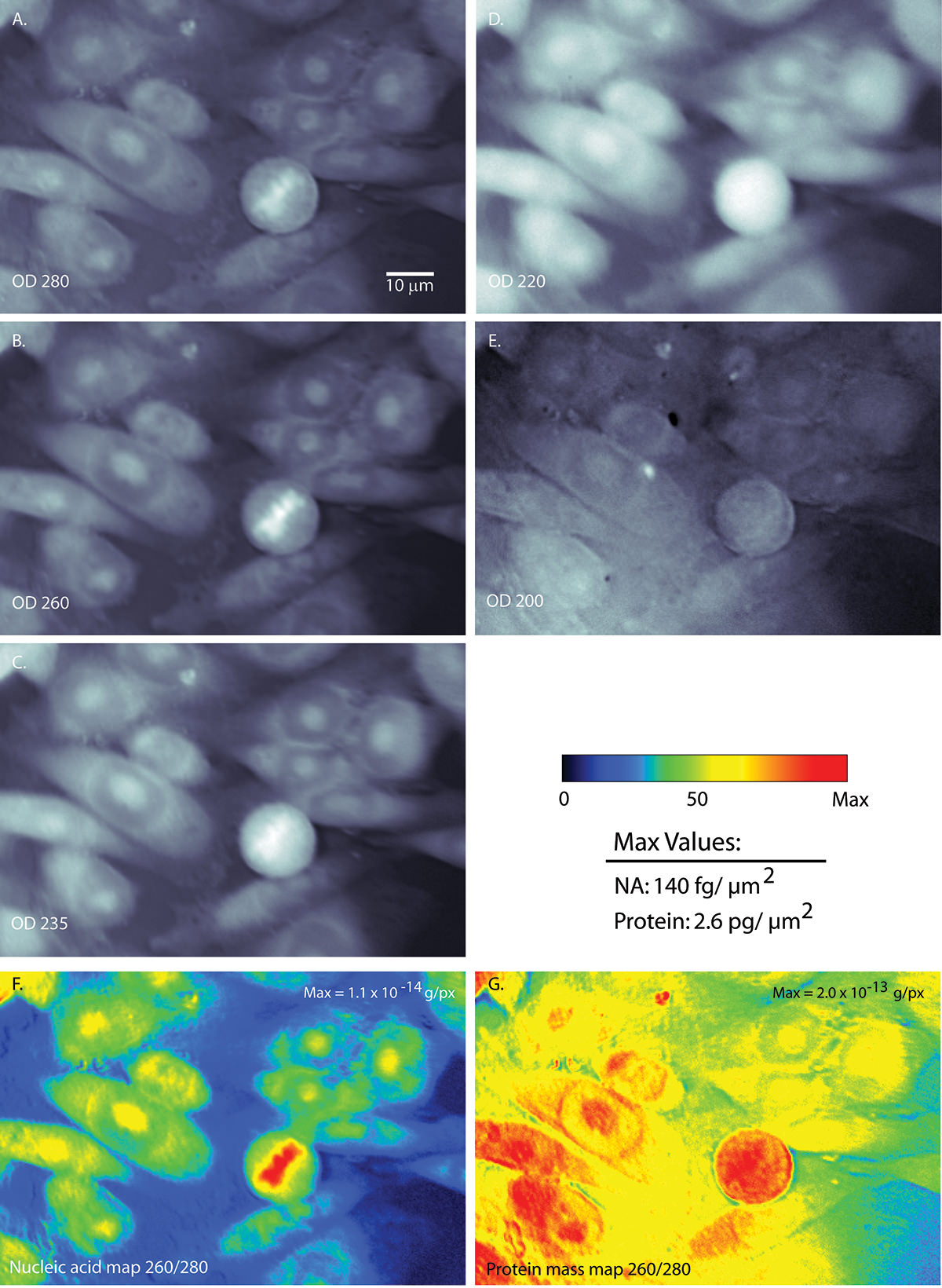
There are seven pictures in the above figure. The grayscale images are the UV images of the same set of cells, at different UV wavelengths. You can see that I imaged the cells at 200 nm, 220 nm, 235 nm, 260 nm, and 280 nm.
With UV, the raw images from the camera, would read out as light areas having been exposed to more UV light. Hence, it also corresponds to areas with less UV absorbed. The images I present are actually maps of optical density; here, white implies higher density and hence, more absorption.
A graduate student, Ben Zeskind, at MIT had implemented this UV microscope. He used LEDs that emitted at 280 or 260nm. These wavelengths have traditionally been used in spectroscopy to identify nucleic acid content in bulk (i.e. in solutions after having ground up and purified the compound from millions of cells.) Naturally, there is value in attempting to do the same, at the level of individual cells.
For any number of reasons, a scientist might be skeptical that there is enough concentration of each material to be apparent. These microscope images are of course 2-dimensional. A cell is actually 3-D. The best way to describe the shape of these cells is literally a sunny-side up egg. It’s flat along the edges and raised where the yolk is. So goes these cells. Luckily for Ben’s project, it turned out there is sufficient material to absorb UV appreciably, within each “column”. UV absorption depends on the sum of all the materials in the light path taken by UV through the cell, on its way to be captured by the pixel. You might easily predict that path of light taken through the nucleus would exhibit more stuff (since it is thicker).
One final trick in the property of UV light is that different materials absorb UV light differently. If we compare pure protein mass with nucleic acid, we see that proteins absorb UV much more strongly than nucleic acids. However, the disparity between the two changes for different wavelengths. At 280 nm, proteins absorb more than 15 times more strongly than nucleic acid. At 260 nm, that difference drops to a 5-fold difference. Ben was able to use that slight difference to generate the mass maps.
What I was able to contribute was to use UV light that was more energetic (200 and 220 nm); in this regime, protein absorbs UV 100 times more strongly than nucleic acid. Using 220 nm is a more optimal method for separating proteins and nucleic acids in a cell. I adapted a way to calculate the protein absorption of UV at 220 nm and applied to our mass map method. Also rather fortunate for us, fats and sugars absorb more strongly than protein at even more energetic wavelengths. In the UV band we are using (220 to 260 nm) the contribution of these other types of molecules was lessened. In the end, I was able to use this technique to look at various things, like nuclei of chicken blood, to determine its nucleic acid content. There had been other methods used to “weigh”, or more properly “mass”, the amount of matter in these structures. Using our method, we measured numbers that are credible and within range of previous measurements.
Again, what does this have to do with the truth?
Remember what I told you; we are using images in a quantitative fashion. I am saying that, if you were able to grow cells on glass (actually, on quartz crystals), fix them, and then image them with a UV microscope, you can actually measure the amount of matter in each cell. I spoke a lot of protein and nucleic acid; how am I supposed to convince you that my method is specific enough to count up either mass, separately?
The first piece of evidence can be seen in the gray images. Compare the top two images, one taken at 280 nm and the other at 220 nm. You can see that there are differences in optical density for a circular cell in the lower part of the field. For the 280 nm image, there is a bit of matter concentrated in the center. For the 220 nm image, that same cell looks bright all the way across. That is evidence showing that the different wavelengths absorb differently. When you look at the mass maps (the colored images on the bottom), you can see something similar. The nucleic acid map shows a concentration of mass in the cell’s center, while the protein map shows mass everywhere. We also know, from the DNA stain I told you about, that the mass in the center of the nucleic acid map is actually where the DNA is located. Thus that is more evidence that the processing Ben used did what it was supposed to do.
I modified the formula to take advantage of 220 nm and 260 nm. We used a new type of UV light source, which allowed us to use shorter UV wavelengths. In the end, I found that 220/260 wavelength pair is an option that gave more consistent results. Using the DNA stain, I was able to write an algorithm to extract the masses of cells stained by DNA label, providing an “objective” way of measuring nucleic acid mass from the images. The objective part comes because it does not rely on humans to identify a cell structure. In this way, we were able to show that the mass of nucleic acid in Chinese Hamster Ovary cells, along with chicken blood nuclei, had the requisite amount.
How about protein? For this, we used human blood; using the same method, we found that our method massed, across many cells, the expected amount of protein.
Why all the alchemy with chicken and human blood? Chicken blood is interesting in that it has nuclei. Not only that, we have estimates of the nucleic acid content derived by biochemistry experiments (such as identifying the number of phosphate molecules in a sample, given that we know how many cells were used to get that sample.) Phosphate is a good proxy because there is one between each base; since DNA is a “ladder” made up of base pairs, there are two phosphate molecules between each base-pair. This known and constant proportion (or, as scientists say, stoichiometry) allows us correlate phosphate to nucleic acid.
Human blood cells (and actually a fair number of other blood types) are without nuclei. That means they are all protein. Correcting for the fact that the proteins have a strongly light absorbent compound in heme, also allows us to use human red blood cells as a calibration marker.
Ultimately, this led us to create a library of cell mass.
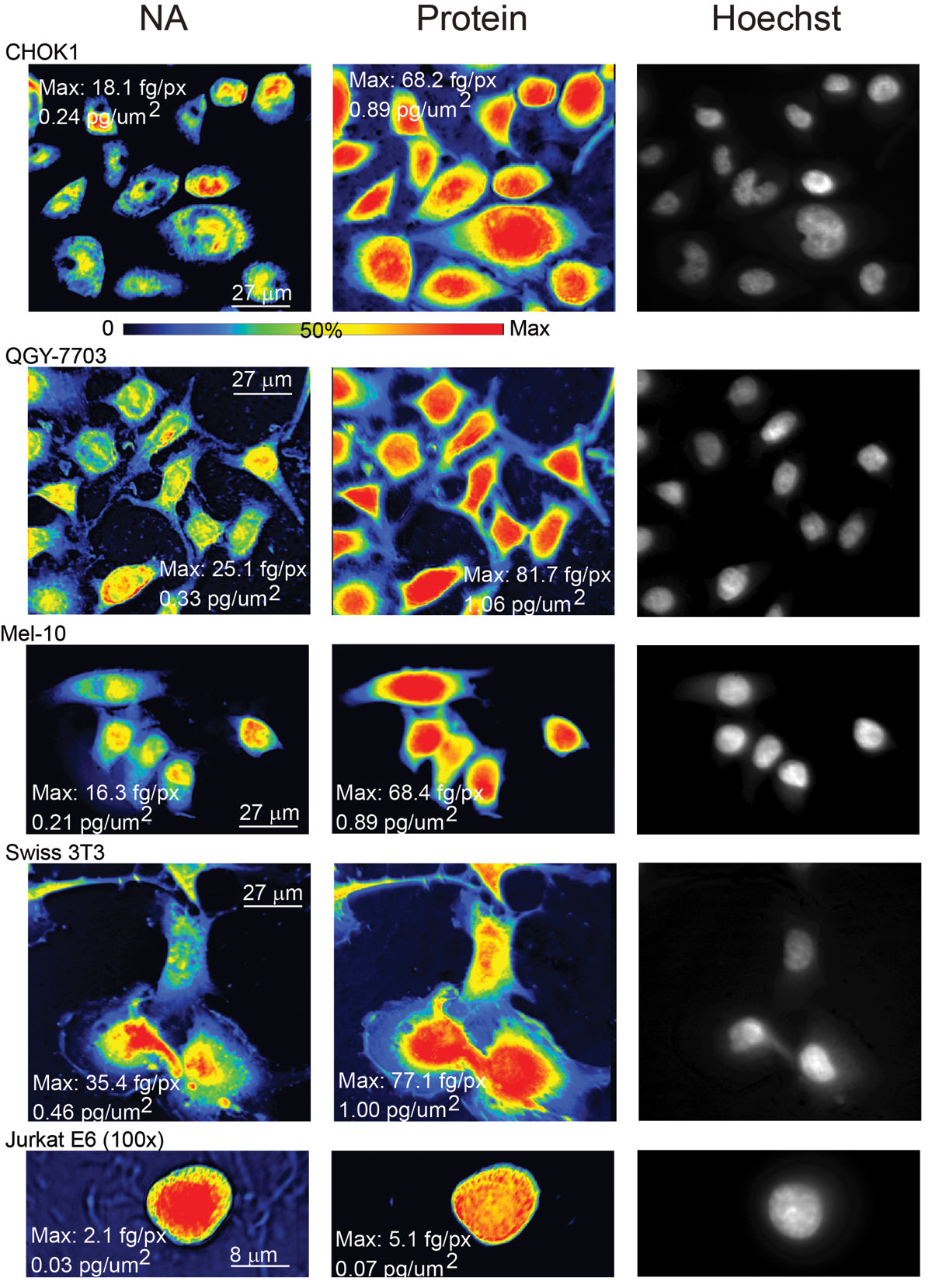
Each row in the above image shows the nucleic acid maps, protein maps, and then an image of the same field of cells with the DNA stain. Each row contains a different cell type. You can see the array of cellular features that characterize each cell, when they are plated out in a single layer. For the record, the rows contain Chinese Hamster Ovary cells, liver cancer cells, a melanoma cell, a fibroblast cell, and a white blood cell.
This was pretty much a whistle-stop tour of about two years worth of research. Photoshop has got nothing on me, as I took images and “manipulated/processed/converted” RAW, 12-bit grayscale numbers into a “mass per pixel” quantity. Simply encircling each cell will allow you to count up the total mass of either protein or nucleic acid.
So I am fairly confident when I say that images can convey truth, or at least something that can be demonstrated repeatedly and reliably, not only by me but by others. I was able to teach undergraduates how to work this microscope. Using their RAW data and my program, they can generate numbers that are consistent within our lab. We were also able to teach another lab, whose members actually imaged protein fibers and determined their mass! They were able to use that to estimate a value for a model of stress in biomolecules. Pretty neat!
In the end, perhaps none of this is controversial to the reader. Of course there is some reality out there. I don’t think anyone really denies that we shouldn’t play near cliffs or point sharp objects at each other.
Then where is the controversy? What people mistake as truth is really their interpretation of an event, and is something they value more than anybody else’s interpretation. Therefore they wish others to agree. The differences between subjectivity and objectivity comes when someone overlays a narrative interpretation over facts. Antagonism arises when people offer different interpretations.
At a basic level, no one can deny that I imaged something. Whether I’ve done enough to address your concerns that I imaged what I said I imaged, and that these images provide a basis for calculating mass, is what’s up for grabs. The most I can do is to place my research into a historical context and to suggest that it allows us to project our knowledge slightly into new territory. I can point to links between older techniques that used UV to determine mass, to the newer experiments comparing my image to known dyes, and to known consequences given the use of cells with only proteins or with a known quantity of nucleic acid.
I could still be wrong, but if I did my job, the error should be subtle and nuanced. Interestingly enough, it isn’t enough to simply show that I am wrong. What settles the issue, against my favor, is for the new guy to show how I went wrong and to explain how I got my observations wrong. This is why scientists can’t gloat too much if they prove someone else wrong. If we all do our best, then a legitimate reason for getting things wrong is simply that our tools or observations weren’t good enough. It’s not a mystery at all where we went wrong. Since technology pretty much improves, so does our accuracy. The only thing I can guarantee is that, over time, what I found will be improved upon. This would be proper functioning of science.
To give a trivial example, no one would deny that Newton gave us a good model of mechanistic gravity. But he was unaware of quantum electrodynamics. When Einstein gave a better, more accurate model of gravity and propagation of electromagnetic waves (such as light) in vacuum, scientists can actually identify where Newton went wrong; his model of gravity is sufficient for slow moving objects. As our speeds approach the speed of light, Newton’s model of gravity and kinetic energy becomes less and less accurate.
It isn’t that one person has a privileged position; at most, the evidence I have presented gives me a slightly higher vantage point. For someone to dislodge them, they need to provide compelling evidence. Interpretation, agreement, and consensus are by-products of this whole process. It isn’t that I don’t have to present my evidence. I already did. It fits into a particular context. People who disagree need to draw on a compelling set of experiments and arguments to first show that I am wrong, and then how I arrived at my errors.
It is easy to see why such a view point is so difficult to apply to the real world. For one thing, we have a lot of emotional, financial, and political stake in promoting specific points of view. And it’s so damned hard to tease apart the complexity of life, in the same way that I had done to the cells.
Likewise, no photograph is without bias, since where one stands is a function of the photographer’s experience, access, and yes, what he or she wishes to show. By the same token, because photographs capture a little slice of time, it is more than a painting or a courtroom sketch. We want photography to be documentary truth, simply because of the way a photograph looks.
From my scientific background, however, a single picture just isn’t enough. Sure, given a story or essay, a single photograph can encapsulate it. Even if a picture is worth a thousand words, I’m not sure a thousand words are enough. You might actually need a few more pieces of evidence.
Likewise, my scientific articles talking about this UV imaging technique does not just depend on the one or two images. For each object type (in my case, cell type or red blood cell type), I imaged hundreds of cells. I had an algorithm that used the nucleic acid stain to isolate the nuclei. At each stage in data collection and analysis, I needed to ensure that I am doing what I documented. The point is that a single image or experiment is rarely enough. I showed you four images, most of which are from the same figure. I haven’t told you the other experiments or analysis I performed, which all addressed one point: that UV microscopy can be used to create mass maps, and from these maps, the matter contained in a cell can be measured. I explored the consequences of what having a UV mass measurement technique means.
However, a single photograph, or a scientific image, does show something happened in front of the camera. Here’s an illustration of my point. We instinctively understand the type of pictures we can get, depending on who is in front of us and behind us. We can be shooting a protest from the side of the protesters, and we expect to see (and it’s becoming more and more apparent even in the USA) police in riot gear, set in a line 10 across and several deep, faceless machines with batons or perhaps tear gas canisters at the ready. If the photographer were embedded with the police, we expect to see hooded men, throwing rocks or Molotov cocktails. They are walking around brazenly with bats, perhaps looking to crack a window for looting the store.
Whether the photographer manipulated the scene (i.e. asked the subjects to pose, rearrange things, used flash, took angles designed to stress fear and threat) matters to the extent of the point he or she wishes to show. To demonstrate an emotion, perhaps 1 frame is sufficient; here, we might want a lot of art to the image. To do a reportage, a few images forming an essay might be needed. To indict the behavior of police or rioters? Eyewitness testimony, reporters on the scene, video cameras, crowd-sourced images and video, and police cameras are the bare minimum amount of information needed. And of course as few tricks as possible. However, much the photojournalist conforms to or ignores these rules, it doesn’t change the fact that policemen were organized into a Roman phalanx and civilians were fighting back.
Clearly, a single point of view is not enough. We would be remiss in our duty as interpreters of images to not ask, what exactly was behind the photographer to elicit this reaction? The question of whose point of view is “the truth” is for the individual to decide if these separate pieces of evidence support one view point over another. But something did happen. And I understand what it means to be on opposite sides of the interpretation divide, for it also means that we are being judged.
What we have to realize is that it takes a level of rigor and certainly thick skin to be able to distance ourselves from the emotion of the situation, to understand that analyzing the image at hand differs from offering an interpretation. The latter simply requires a lot more information. You don’t need complete information, but you need to be able to string together independent streams of evidence to build a narrative. Is the interpretation the right one? Is it the truth? Therein lies the difference between knowledge and wisdom. Knowledge might tell us who did what to whom. Wisdom allows us to step back and judge whether we have used the best data and arguments, and whether we are willing to excuse or support some behavior. This seems like it’s beyond the scope of any one photographer, let alone a picture or even a photoessay.
This guest post was submitted by Dr. Man Ching Cheung, a clinical engineer working at Bringham and Women’s Hospital. You can see more of Dr. Man Ching Cheung’s work on his 500px page.
Sorry TL;DR ;)
Ok, to be true to the truth ;) I scanned it quickly … very deep article, but just as another reader said… it’s a bit dense for this website, even more so if it actually just wants to say “the truth is everyone’s own truth and doesn’t always coincide with anyone elses”
Anyway, since it doesn’t take up space (well a few KBytes) and we should always be thankful for high quality information of any kind, thanks for the efforts!
I’m sure that this is a great article , but I don’t know what is doing in this blog.
Wow, what a great piece Dr. Cheung! As an undergraduate student on astronomy I must recognize that you got me more with the description of your work than with the message of your article (which by the way I completely agree, and as a form of recognition I view my photography purely as a way of expression), as it is intellectually interesting reading about the other end of imaging (as we do it for the vastness of space), and reminded me of a lecture of one of the professors of my faculty who work on quantum optics about optical tweezers (but that would be all that I recall right now).
By the way, nice work on your more artistic side that share on 500px.
What a great, informative article. I’ve been looking to learn more about photographing microscopic subjects and this is just great. Thanks, Dr. Cheung!
Interesting article. Thanks. There is no real objective ‘truth’ in any art or discipline, including photography. Even a representation of landscape, which seems to be de rigeur for this site, is in doubt. I’m currently reading “Land Matters” by Liz Wells, a book written on the tradition of landscape photography, in which she establishes that the act of observation changes the observed. I’ve simplified what she’s getting at for the sake of brevity, but I think that it’s a reasonable precis. There is much philosophical thought behind a photograph, many filters and preconceived ideas which effect our ways of observation. You might find “The Poetics of Space” by Gaston Bachelard an insightful read too. I’m reading these as part of my research into landscape for my post grad studies in photography.
Stephen – for whatever it’s worth, I’d be VERY interested in anything you write as part of those post-grad studies along these lines. Those themes are all ones I identify closely with – will check out Wells and Bachelard!
Hi Christopher
Thanks, I feel I should warn you that my thesis will be almost totally subjective – I wanted it that way, and will involve research and imagery/photography toward a “response to place using abstractions and symbols”, which is why I’m reading Wells and Bachelard. The “place” could be anywhere, but I’m using landscape for a long list of reasons. Liz Wells really is a prolific and interesting visual arts writer whose history is with photography. It’s pretty exciting stuff. I’m currently researching for a class paper, and will start writing once that’s finished.
Not sure what would be the best way to go about sharing my writing with you, but if you have any ideas let me know, I’d be happy to.
I will just add that scientists can go further with state-of-the art microscopy techniques nowadays and even look at a single molecule (e.g. a protein) in a single cell. And so you can for example direcly count single proteins and follow walkabouts of each in a cell. And you can do it in real time in a living cell, rather then in fixed dead cell. This is all thanks to superresolution techniques that earned Chemistry Nobel this year.
Absolutely, Lukasz! As you noted, it is fantastic to be able to follow a the “life” of a single molecule as it goes about its business. Another important technique is use of structured illumination and deconvolution algorithms to perform single molecule imaging and that also allows for “supraresolution” microscopy. Supraresolution microscopy can resolve structures sized below the diffraction limit of 0.2 micrometer.
Back to the single-protein labeling: the trick is in the counting, of course. Given the average mass of proteins in a cell is approximately 200 picograms, and the molar mass of an “average” protein is about 160 000 g/mol, there are approximately a few hundred million proteins in the cell.
Photographers note that one needs the right tools for the job. The scientific “job” depends on the nature of the question, and there is some utility in having a technique to see a single molecule and another technique to “weigh” proteins in the aggregate. And of course, that differs from a purely bulk method to assess mass in ground up samples.
However, we did try and succeeded imaging live cells under UV microscopy. For reasons of time and due to our particular questions, we did not pursue this to the fullest. In part, there were some difficulties in growing cells on quartz glass. We followed a few cells for several hours: the limited throughput would have precluded us examining hundreds of cells for many cell samples and experiments.
There would have been another set of experiments to demonstrate that cells can survive UV exposure over the timeframe of our experiments. We had doubts that cells can survive over 48 hours, with multiple UV exposures to generate maps of mass redistribution with enough temporal resolution (ideally, we would have liked something around 20 minutes between each set of exposures, for the entire duration of the experiment.)
It is also unclear what the utility of that measurement would be: the whole of science has about looking at higher resolution, at more precise things. As you noted, single molecule imaging not only has the advantage of resolution but we would have made a specific label for it. That is, it is an identified molecule. Our method would have been a rough estimate of what is happening to protein, and only if we can detect changes in bulk mass distribution in the cell.
I hope this adds a bit more context that is useful to you!
mcc
It is just superresolution microscopy, PALM and STORM are mostly used. I am not saying you would need it in your study, I am just generally saying that when it comes to optical (fluorescence) microscopy science has already gone beyond diffraction limit and you can easily image cellular structures at 0.02 micrometer resolution instead of conventional (textbook) limit of 0.2 micrometer resolution (superresoution imaging comes at the cost of sacrificing time resolution tough). The obvoius advantage of supperresolution imaging is that you can now resolve cellular structures much better, literally see 10x better inside the cell than you were able to do before. These same supperresolution techiques (PALM and STORM) allow for tracking motions of signle molecules in a living cell. This on the other hand is important becuse we simply want to study biology of the cell and so it better be alive during our measurements (I admit I am a bit sceptical about using UV ilumination with live cells whose physiology one want to study). Because you can precisely measure the motion (diffusion) of a single molecule in a cell you can tell when it is moving faster, or slower, or it stays still, indicating the molecule moves freely throughout different cell’s areas or it is temporairly bound to some other molecule/s or cell components (so you in fact study the function of this molecule in a real time). Superresolution methods also allow for counting to the single molecule in a cell (of course you need to know the exact photophysics behind your fluoresent dye or fluorescent protein to pull it off). Good job with the article, thanks to you science found its way to this portal. I just wish the story was a little less technical, I am just affraid it may be a little intimidating to general audience here, but I hope I am wrong.